

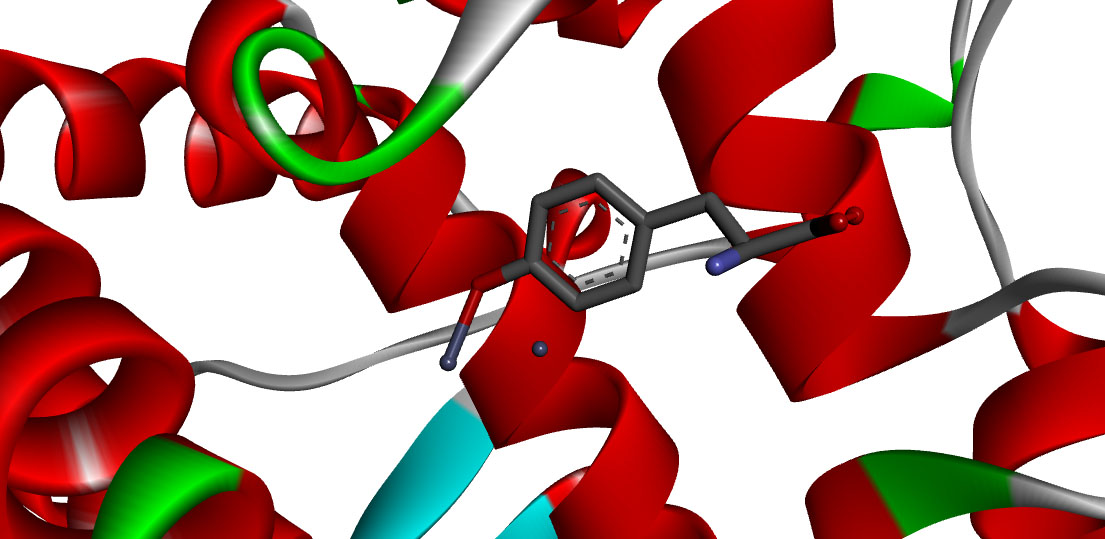
Molecular docking study of tyrosinase inhibitors using ArgusLab 4.0.1 : A comparative study
Abstract
Full Text:
21-25:PDFReferences
. N.C. Cohen. Guidebook on molecular modeling in drug design, 1st Edn. Academic press. Xiii, 1996.
. D.B. Kitchen, H. Decornez, J.R. Furr, and J. Bajorath. Docking and scoring in virtual screening for drug discovery: methods and applications. Nature. Rev. Drug. Disc. 3:935-949 (2004).
. M.A. Thompson. Molecular docking using ArgusLab, an efficient shape-based search algorithm and the AScoring function. ACS meeting Philadelphia 172, CINF 42, PA. (2004).
. A. Hafeez, Z.S. Saify, A. Naz, F. Yasmin, and N. Akhtar. Molecular docking study on the interaction of riboflavin (Vitamin B2) and cyanocobalamine (Vitamin B12) coenzyme. J. Comp. Med. Volume 2013, Article ID 312183: 5 pages (2013).
. A. Naz, K. Bano, F. Bano, N.A. Ghafoor, and N. Akhtar.. Conformational analysis (geometry optimization) of nucleosidic antitumor antibiotic showdomycin by ArgusLab 4 software. Pak. J. Pharm. Sci. 22;1:78-82 (2009).
. A. Oda, and O. Takahashi. Validation of ArgusLab efficiencies for binding free energy calculations. Chem-Bio. Inform. J. 9:52-61 (2009).
. A. Vontzalidou, G. Zoidis, E. Chaita, M. Makropoulou, N. Aligiannis, G. Lambrinidis, E. Mikro, and A. Skaltsounis. Design, synthesis and molecular simulation of dihydrostilbene derivatives as potent tyrosinase inhibitors. Bioorg. Med. Chem. Lett. 22: 5523-5526 (2012).
. M. Sendovski, M. Kanteev, V. B. Shuster, N. Adir, and A. Fisherman. Crystal structure of tyrosinase from Bacillus magaterium in complex with inhibitor kojic acid. J. Mol. Biol. 405:227-237 (2011).
. S. Song, H. Lee, Y. Jin, Y.M. Ha, S. Bae, H.Y. Chung, and H. Suh. Synthesis of hydroxyl-substituted phenyl-naphthalenes as inhibitors of tyrosinase. Bioorg. Med. Chem. Lett. 17: 461-464 (2007).
. Y.M. Song, M.H. Young, J. Kim, K.W. Chung, Y. Uehara, K.J. Lee, P. Chun, Y. Byun, H.Y. Chung, and H.R. Moon. Synthesis of novel azo-resveratrol, azo-oxyresveratrol and their derivatives as potent tyrosinase inhibitors. Bioorg. Med. Chem. Lett. 22: 7451-7455 (2012).
. K. Likhitwitayawuid, A. Sornsute, B. Sritularak, and P. Ploypradith. Chemical transformations of oxyresveratrol (trans-2,4,3ʹ,5ʹ-tetrahydroxystilbene) into a potent tyrosinase inhibitor and a strong cytotoxic agent. Bioorg. Med. Chem. Lett. 16: 5650-5653 (2006).
. M.D. Hanwell, D.E. Curtis, D.C. Lonie, T. Vandermeersch, E. Zurek, and G.R. Hutchinson. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics. 4: 17 (2012).
. N. Jongkon, and P. Tangyuenyongwatana. Molecular binding modes of diarylheptanoids from Curcuma comosa on the ER-β receptor. Thai. J. Pharm. Sci. 38: 82-89 (2014).
. M. Goldfeder, M. Kanteev, N. Adir, A. Fishman. Determination of tyrosinase substrate-binding modes reveals mechanistics between type-3 copper proteins. Nat. Commun. 5: 4505-4509 (2014).
Refbacks
- There are currently no refbacks.